Please click here to order lab tests.
For more information, please also refer to the UCSF Benioff Children’s Hospital Oakland lab menu:
https://www.testmenu.com/UBCHOLab
Clinical Diagnostic Testing is performed under CLIA and CAP licensure and includes assays for routine hemoglobinopathy screening as well as molecular methods for diagnosis of hemoglobin disorders, including sickle cell disease, beta thalassemia, Hb H disease, as well as specialized tests for unstable hemoglobins and other hemoglobin disorders.
Description of Routine Hemoglobinopathy Assays:
Cellulose acetate electrophoresis (CAE) at alkaline pH (8.2 to 8.6) permits separation of the major hemoglobins (Hb A, Hb F, Hb S/D, Hb C/E/O-Arab, Hb G-Philadelphia) based on the relative mobilities of variant hemoglobins. Hemoglobin bands are visualized by staining with amido black and acid violet (or similar stains).
Citrate agar acid electrophoresis at acid pH (6.0 to 6.2) permits separation of the major hemoglobins based on the relative mobilities of variant hemoglobins: Hb C from Hb E and Hb O, as well as Hb S from Hb D and Hb G. However, electrophoresis will not distinguish Hb E from Hb O, nor Hb D from Hb G. Hemoglobin bands are visualized by staining with amido black and acid violet (or similar stains).
Isoelectric focusing (IEF) on agarose gels separates hemoglobin (Hb) fractions and variants based on their isoelectric points. Hb A and Hb F are clearly resolved by this method. Hb C can also be distinguished from Hb E and Hb O, and Hb S can be distinghuished from Hb D and Hb G.

The Free Erythrocyte Protoporphyrin (FEP) test measures the non-complexed, non-heme protoporphyrin concentration in blood. This test alone is not sensitive enough to diagnose iron deficiency, but an elevated FEP in conjunction with a low MCV is suggestive.
FEP is measured using a thin smear of whole blood placed on a coverslip. The fluorometer measures the amount of free unbound porphyrin which accumulates in iron deficiency as a result of decreased heme synthesis. The intensity of the analytic signal produced by the instrument is a function of the molar ratio of fluorescing protoporphyrin to light-absorbing heme.
The Sickle Solubility Test (SST) The Sickle Solubility Test (SST) is used to screen for the presence of sickling hemoglobins. A positive result must be confirmed by another method (HPLC or electrophoresis) to confirm the presence of Hb S and to distinguish Hb AS (carrier state) from Hb SS (sickle cell disease). Other insoluble hemoglobins, such as Hb C-Harlem, will also give a positive result. The SST utilizes a procedure based upon phosphate solubility whereby erythrocytes are lysed by saponin and the released hemoglobin is reduced by sodium hydrosulfite in a phosphate buffer. Reduced HbS is characterized by its very low solubility and the formation of neumatic liquid crystals (tactoids). The resulting tactoids of HbS or non-sickling hemoglobin (e.g. HbC-Harlem) causes the solution to remain turbid. The presence of HbA under these same conditions results in a clear red solution.
Counts of pitted red blood cells (RBC) can be used to assess splenic reticuloendothelial function in patients with sickle cell disease. There is an inverse relationship between the pit count and the degree of splenic dysfunction. Less than 3% of RBCs contain pits in individuals with normal splenic function, whereas up to 50% of RBCs may demonstrate pitting in individuals with functional asplenia.
Erythrocytes are fixed with 1% glutaraldeyde and examined under phase contrast microscopy to visualize and manually count pitting as an estimate of the number of pitted red cells.
For collection, visit: https://www.testmenu.com/UBCHOLab/Tests/924899
References:
Marsh WL Jr, Nelson DP, & Koenig HM: Free erythrocyte protoporphyrin (FEP) II. The FEP test is clinically useful in classifying microcytic RBC disorders in adults. Am J Clin Pathol 1983; 79(6):661-666
Nalbandian RM, et al. Dithionite tube test: A rapid, inexpensive technique for the detection of hemoglobin S and non-S sickling hemoglobin. Clin Chem 1971; 17:1028-1032
Fatunde, O.J. and Scott, R.B. 1986 J Ped Hem Onc
Witkowska, HE, et al. Sickle Cell Disease in a Patient with Sickle Cell Trait and Compound Heterozygosity for Hemoglobin S and Hemoglobin Quebed-CHORI. NEJM. 1991. Vol. 325 No. 16. Pp 1150-1154.
Allele-specific assays are used to target the presence or absence of a specific allele. Allele-specific PCR employs a set of manufactured oligonucleotide primers that hybridize to and specifically amplify sequences of DNA containing the target alleles of interest (eg, Hb A, Hb S, Hb C, Hb D, Hb E and Hb O-Arab). Analysis of the PCR products generated can be done by electrophoresis based on the length of the fragments generated.
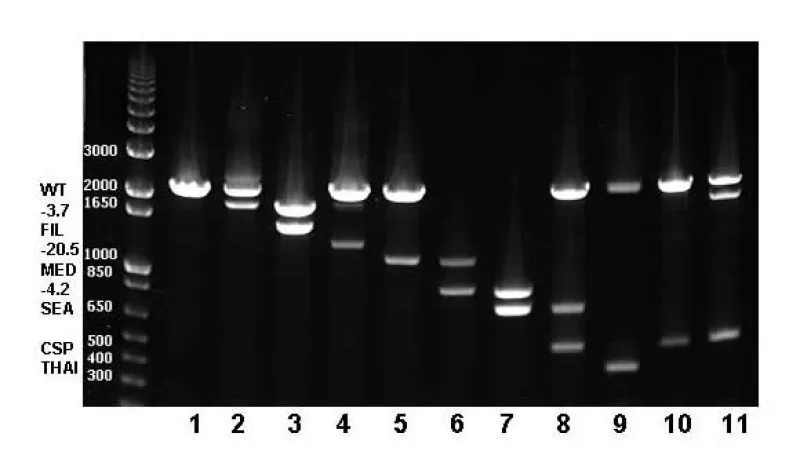
Multiplex Gap PCR techniques (amplification using oligo-primers flanking deletion breakpoints) are used to detect common alpha thalassemia deletion mutations, alpha gene duplication, and other globin gene deletions, such as Hb Lepore and HPFH.
Gap PCR is based upon the inability of PCR primers complementary to DNA sequences that are far apart to direct amplification unless a deletion brings them closer together. PCR primer pairs are designed to flank a known deletion, generating a unique amplicon that will be smaller in the mutant sequence compared with the wild type. The presence or absence of PCR product is detected by electrophoresis. Primers specific for 7 of the most common alpha thalassemia deletions, as well as the CS point mutation are multiplexed to detect the mutations most often responsible for HbH disease. This technique is also used to detect the delta/beta globin gene crossover responsible for Hb Lepore and the large deletions responsible for hereditary persistence of fetal hemoglobin.
Alpha thalassemia deletions (and CS)
Alpha gene duplications
Delta-beta deletions (HPFH-1, HPFH-2, HPFH-3, HPFH-7, Lepore)
Beta-globin deletions (beta-FL, -619bp, Asian-Indian inversion)
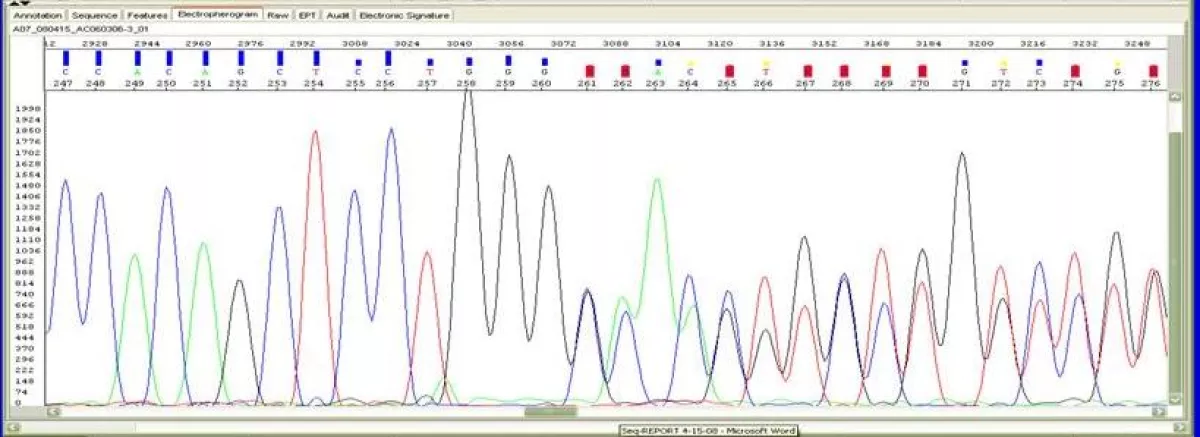
DNA Sequencing analysis of alpha, beta or gamma globin genes is performed to definitively identify unknown point mutations or sequence variation. DNA sequencing for globin gene mutations is performed on the Applied Biosystems Model 3130XL Genetic Analyzer. Genomic DNA samples submitted for sequencing and automated analysis undergo a cycle sequencing reaction. The fluorescently labeled products are separated from reaction reagents and analyzed by capillary electrophoresis. The DNA sequence is automatically analyzed by software. Typical read lengths are 600 - 650 bases for high quality DNA templates and primers. Sequencing results are reviewed manually for overall quality, and accuracy of the base call. Whenever necessary, the samples are either reanalyzed with a different base caller and the bases edited or reloaded to improve the quality.
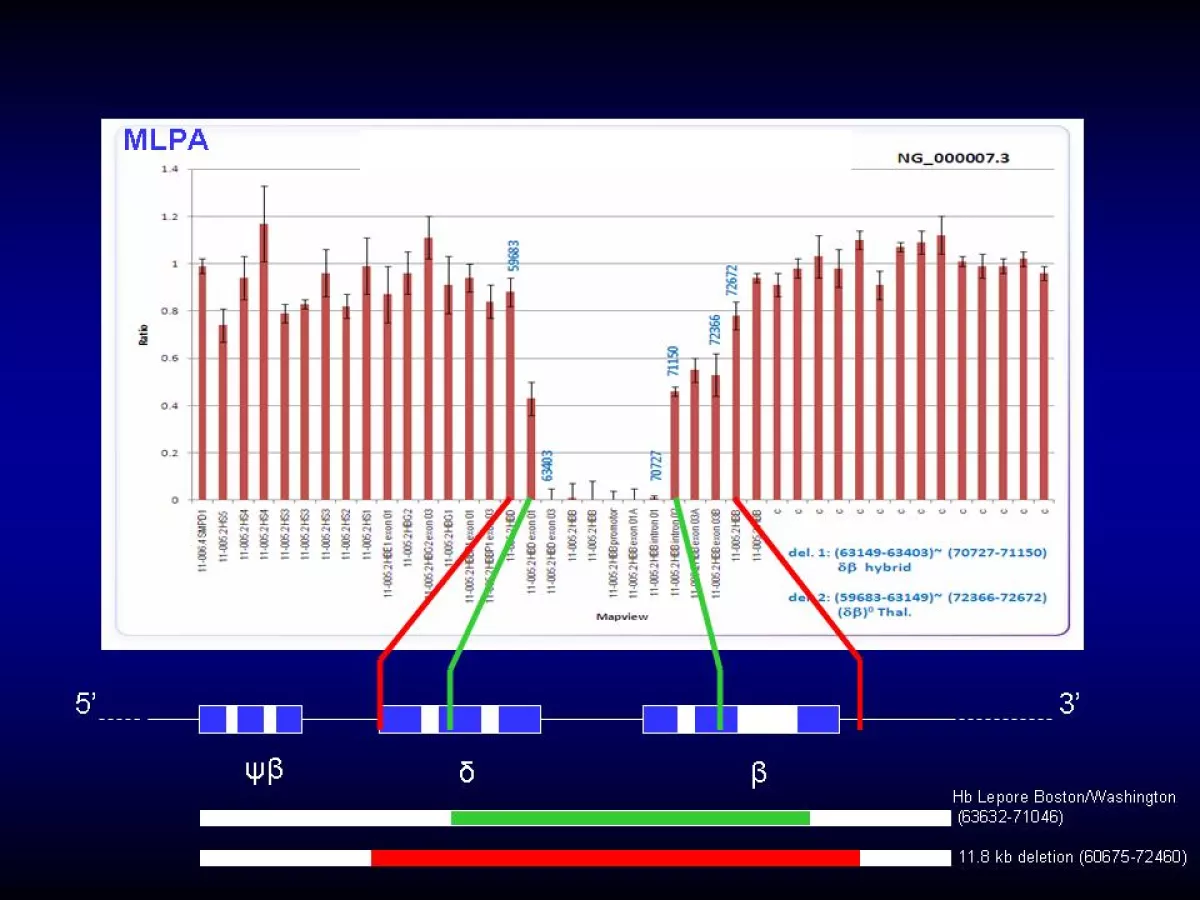
Multiplex Ligation-Dependent Probe Amplification (MLPA) is used to identify large or unknown alpha-thalasssemia or beta-thalassemia deletions. Each target DNA sequence has a pair of specific probes that contain a unique length stuffer sequence so that each probe pair can be distinguished based on its length. If the target sequence is normal, the probes will hybridize with the target and are then ligated by a ligase. These ligation products are amplified using only one primer pair and the products separated by capillary electrophoresis. The amplification product of each target will have a a unique length permitting separation using a sequence analyzer. The relative amounts of probe amplification products correspond to the relative copy number of target sequences. A 50% reduction in PCR product indicates a heterozygous deletion, while complete absence of amplified product indicates a homozygous deletion. MLPA can only determine the extent of the deletion and the approximate location of breakpoints. DNA sequencing is subsequently performed to define the deletion breakpoints.
Special Hb Tests:
Heat stability Test (HST) for Unstable Hemoglobins is performed for detection of unstable hemoglobins or hemoglobins with altered oxygen affinity. Some hemoglobin mutations result in an unstable hemoglobin that has decreased solubility and precipitates to form erythrocyte inclusions (Heinz bodies). These inclusions reduce the deformability of the red blood cell and lead to hemolysis. Examples of unstable hemoglobins are Hb Gun Hill, Hb Leiden, and Hb Köln. Hemolysate is warmed at 50°C for one hour. Unstable hemoglobins will precipitate under these conditions and can be visually identified.
Isopropanol Test (IT) for Unstable Hemoglobins is performed for detection of unstable hemoglobins or hemoglobins with altered oxygen affinity. Some hemoglobin mutations result in an unstable hemoglobin that has decreased solubility and precipitates to form erythrocyte inclusions (Heinz bodies). These inclusions reduce the deformability of the red blood cell and lead to hemolysis. Examples of unstable hemoglobins are Hb Gun Hill, Hb Leiden, and Hb Köln. Hemolysate is incubated in a Tris buffer containing isopropanol. The less polar isopropanol buffer stresses the sulfhydryl bonds of the Hb molecule. Normal hemoglobins will not precipitate within 20min, while unstable hemoglobins will precipitate within 5 minutes.
Hepcidin Immunoassay The Intrinsic Hepcidin is an ELISA test for the quantitative measurement of Hepcidin in human serum and plasma. Hepcidin is a peptide hormone that regulates dietary iron absorption from the duodenum, controls recycling of senescent erythrocyte iron by macrophages, and manages iron transport from hepatocytes into plasma. Hepcidin is positively regulated by plasma iron and IL-6 (inflammation, infection) and is suppressed by erythropoiesis via erythroferrone. Abnormally low serum hepcidin is associated with iron deficiency anemia (IDA) and hereditary hemochromatosis, and high serum hepcidin can lead to iron sequestration and to anemia of inflammation (anemia of chronic disease) observed in chronic kidney disease (CKD), rheumatoid arthritis (RA), various cancers, and iron refractory iron deficiency anemia (IRIDA). Hepcidin levels have been shown to be useful in predicting response to oral iron therapy.
For collection information, visit: https://www.testmenu.com/UBCHOLab/Tests/1061714